
Team 4 - Thierry Tordjmann
Biliary homeostasis and liver repair
Research activities
1. Biliary homeostasis and liver repair
Supervision: Dr Thierry Tordjmann
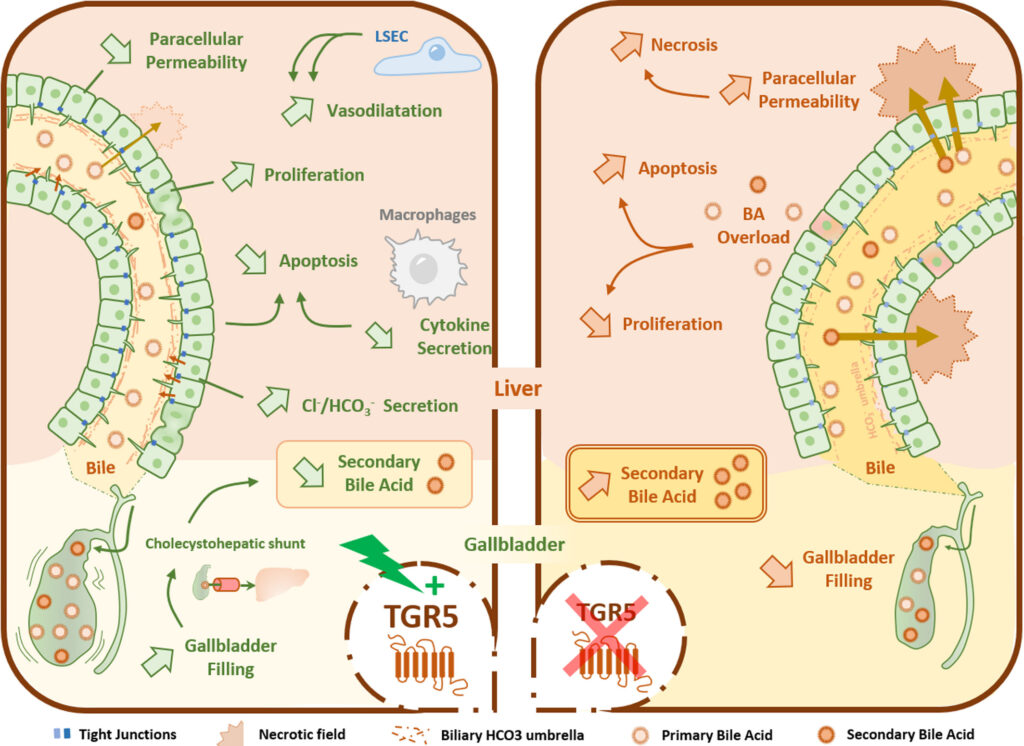
Even though liver regeneration capacity is huge, tissue repair during chronic injury (biliary or non-biliary etiologies) can lead to hepatic fibrosis. Importantly, after injury and during liver repair, liver functions have to be maintained to fulfill the peripheral demand. This is particularly critical for bile secretion, which has to be finely tuned in order to preserve liver parenchyma from BA-induced injury. However, mechanisms allowing the liver to maintain biliary homeostasis during repair after injury are not completely understood. Our studies are focused on mutual relationships between biliary homeostasis and liver repair. Particular attention is paid to BA and their receptors (TGR5) as potential targets at crucial regulatory crossroads. To this end, we handle in vivo murine models of experimental BA overload, in vitro studies of hepatocytes and cholangiocytes, as well as human patients liver samples (cells and tissues) analysis.
Figure: Schematic representation of the different TGR5-mediated mechanisms of hepato-protection. The integrity of the blood-biliary barrier (BBB), which separates blood from bile, is essential in both physiological and pathological conditions. TGR5 stimulation promotes: - cholangiocyte proliferation and - barrier function by reinforcing cholangiocyte tight junctions. TGR5 regulates chloride and bicarbonate transport in bile, reducing BA protonation and protecting liver parenchyma from BA cytotoxicity. TGR5 have physiological impact on GB functions, possibly including BA reabsorption reported as the cholecysto-hepatic shunt, restricting the amount of toxic secondary BA. TGR5-mediated BA signalling reduces hepatic vascular tone and portal pressure, resulting from sinusoidal vasodilatation (adapted from Merlen et al., Liver Int 2020;40(5):1005-1015).
2. Pathophysiology and treatment of genetic cholestasis
Supervision: Pr Emmanuel Jacquemin
Chronic cholestatic liver diseases can have genetic origins, due to molecular defects of ABC transporters localized at the canalicular membrane of hepatocytes. Indeed, genetic variations of ABCB11/BSEP (bile acid transporter) and ABCB4/MDR3 (phospholipid transporter) are implicated in rare and severe forms of genetic cholestasis named progressive familial intrahepatic cholestasis types 2 and 3, respectively. These diseases are characterized by chronic cholestasis, icterus and moderate to severe pruritus, and they mostly evolve to liver failure, thus requiring liver transplantation. The aim of our studies is to identify new molecules able to correct the molecular defects of the mutated canalicular ABC transporters (expression, traffic or function), in the frame of genetic cholestasis. As a first line of research, we use cell models expressing the defective transporters in order to identify molecules able to rescue the defective canalicular ABC transporters. Then, we will validate the best molecules at a preclinical stage in humanized mouse models mimicking the human pathologies (KO mice expressing the mutated human canalicular transporters by viral infection) before considering their valorisation and transfer to the clinic.
Figure: Schematic representation of the classes of genetic variants of canalicular ABC transporters. The classes were determined on the basis of the deficit identified in cell models. Class I corresponds to nonsense variants (premature stop codons) leading to extremely reduced or even no protein expression. Class II, III and IV variants are mainly missense variants (replacement of one amino acid by another) whose deficiency is linked to defects in intracellular trafficking, activity or stability, respectively. Class V variants have no apparent defect, identifiable by the experimental approaches used and could correspond to non-deleterious polymorphisms (adapted from Vauthier et al., Biochem Pharmacol 2017;136:1-11).
3. Cellular mechanisms of ciliopathies and biliary organogenesis
Supervision: Dr Pascale Dupuis-Williams
The construction during embryogenesis and the maintenance of a functional biliary system rely on the role of the primary cilium, as exemplified by the biliary diseases associated to ciliopathies. Although much work has helped to elucidate the role of the cholangiocyte primary cilium in signaling and in regulating bile homeostasis, its function in biliary organogenesis remains to be elucidated. In this context, our group focuses on Ciliopathies and Biliary Organogenesis. Its activities are developed according to two axes: i) a research program intended to decipher the role in cholangiocytes epithelial polarity of ciliary proteins involved in bile dysgenesis and ii) a bioengineering activity aiming in the construction of biliary tubes to provide models of in vitro biliary development and for regenerative medicine. in vitro et des outils pour la médecine régénérative.
Team members
RESEARCHERS AND LECTURERS

Olivier DELLIS
Associate professor
Université Paris-Saclay
01-69-15-49-59
olivier.dellis@universite-paris-saclay.fr

Pascale DUPUIS-WILLIAMS
Associate professor
ESPCI Paris
01-69-15-68-66
pascale.dupuis-williams@universite-paris-saclay.fr

Thomas FALGUIERES
Research Associate
Inserm
01-69-15-62-94
thomas.falguieres@inserm.fr

Grégory MERLEN
Research Associate
Inserm
01-69-15-68-59
gregory.merlen@inserm.fr

Thierry TORDJMANN
Research Director
Inserm
01-69-15-68-63
thierry.tordjmann@universite-paris-saclay.fr
PHYSICIANS
Oanez ACKERMANN
Paediatric hepatologist
AP-HP
01-45-21-31-67
oanez.ackermann@aphp.fr

Marion ALMES
Assistant professor
AP-HP / Université-Paris-Saclay
01-45-21-31-67
marionflorence.almes@aphp.fr

Emmanuel GONZALES
Full professor
AP-HP / Université-Paris-Saclay
01-45-21-31-67
emmanuel.gonzales@aphp.fr
Dalila HABES
Paediatric hepatologist
AP-HP
01-45-21-31-67
dalila.habes@aphp.fr
Bogdan HERMEZIU
Paediatric hepatologist
AP-HP
01-45-21-31-67
bogdan.hermeziu@aphp.fr

Emmanuel JACQUEMIN
Full professor
AP-HP / Université-Paris-Saclay
01-45-21-31-67
emmanuel.jacquemin@aphp.fr
Anne SPRAUL
Paediatric hepatologist
AP-HP
01-45-21-35-22
anne.spraul@aphp.fr
Alice THEBAUT
Paediatric hepatologist
AP-HP
01-45-21-31-67
alice.thebaut@aphp.fr
ENGINEERS AND TECHNICIANS

Manon BANET
Study engineer (contract)
Inserm (ANR)
01-69-15-62-94
manon.banet@inserm.fr

Latifa BOUZHIR
Study engineer
Inserm
01-69-15-68-56
latifa.bouzhir@universite-paris-saclay.fr

Khadidiatou DIARRA
Technical assistant
Inserm
01-69-15-68-58
khadidiatou.diarra@inserm.fr

Isabelle DOIGNON
Study engineer
Inserm
01-69-15-68-64
isabelle.doignon@universite-paris-saclay.fr

Olivier FAYOL
Technician
Université Paris-Saclay
01-69-15-63-96
olivier.fayol@universite-paris-saclay.fr

Isabelle GARCIN
Study engineer
Université Paris-Saclay
01-69-15-76-39
isabelle.garcin@universite-paris-saclay.fr

Martine LAPALUS
Research engineer
Inserm
01-69-15-39-25
martine.lapalus@universite-paris-saclay.fr
PHD STUDENTS AND POST-DOCS

Chloé CAENEN
PhD student
Université Paris-Saclay
01-69-15-58-24
chloe.caenen@inserm.fr

Mounia LAKLI
PhD student
Université Paris-Saclay
01-69-15-62-94
mounia.lakli@inserm.fr

Thibault PEBRIER
PhD student
Université Paris-Saclay
01-69-15-63-96
thibault.pebrier@universite-paris-saclay.fr
ALUMNI
XXX XXXXX
XXXX
XXXXXXXXXX
01-XX-XX-XX-XX
xxxx.xxxxx@xxxx.com
XXX XXXXX
XXXX
XXXXXXXXXX
01-XX-XX-XX-XX
xxxx.xxxxx@xxxx.com
XXX XXXXX
XXXX
XXXXXXXXXX
01-XX-XX-XX-XX
xxxx.xxxxx@xxxx.com
Main publications
2022
- In Vitro Rescue of the Bile Acid Transport Function of ABCB11 Variants by CFTR Potentiators. Mareux E, Lapalus M, Ben Saad A, Zelli R, Lakli M, Riahi Y, Almes M, Banet M, Callebaut I, Decout JL, Falguières T, Jacquemin E, Gonzales E. Int J Mol Sci. 2022 Sept 15;23(18):10758. doi:10.3390/ijms231810758.
- ABC Transporters in Human Diseases: Future Directions and Therapeutic Perspectives. Falguières T. Int J Mol Sci. 2022 Apr 12;23(8):4250. doi: 10.3390/ijms23084250. PMID: 35457067.
2021
- Construction of functional biliary epithelial branched networks with predefined geometry using digital light stereolithography. Mazari-Arrighi E, Ayollo D, Farhat W, Marret A, Gontran E, Dupuis-Williams P, Larghero J, Chatelain F, Fuchs A. Biomaterials. 2021 Dec;279:121207. doi: 10.1016/j.biomaterials.2021.121207. PMID: 34741977.
- Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): a randomised phase 2 study. Gonzales E, Hardikar W, Stormon M, Baker A, Hierro L, Gliwicz D, Lacaille F, Lachaux A, Sturm E, Setchell KDR, Kennedy C, Dorenbaum A, Steinmetz J, Desai NK, Wardle AJ, Garner W, Vig P, Jaecklin T, Sokal EM, Jacquemin E. Lancet. 2021 Oct 30;398(10311):1581-1592. doi: 10.1016/S0140-6736(21)01256-3. PMID: 34755627.
- In vitro functional rescue by ivacaftor of an ABCB11 variant involved in PFIC2 and intrahepatic cholestasis of pregnancy. Mareux E, Lapalus M, Ben-Saad A, Callebaut I, Falguières T, Gonzales E, Jacquemin E. Orphanet J Rare Dis. 2021 Nov 18;16(1):484. doi: 10.1186/s13023-021-02125-4. PMID: 34794484.
- Self-Organogenesis from 2D Micropatterns to 3D Biomimetic Biliary Trees. Gontran E, Loarca L, El Kassis C, Bouzhir L, Ayollo D, Mazari-Arrighi E, Fuchs A, Dupuis-Williams P. Bioengineering (Basel). 2021 Aug 5;8(8):112. doi: 10.3390/bioengineering8080112. PMID: 34436115.
- RAB10 Interacts with ABCB4 and Regulates Its Intracellular Traffic. Ben Saad A, Vauthier V, Lapalus M, Mareux E, Bennana E, Durand-Schneider AM, Bruneau A, Delaunay JL, Gonzales E, Housset C, Aït-Slimane T, Guillonneau F, Jacquemin E, Falguières T. Int J Mol Sci. 2021 Jun 30;22(13):7087. doi: 10.3390/ijms22137087. PMID: 34209301.
- Molecular Regulation of Canalicular ABC Transporters. Ben Saad A, Bruneau A, Mareux E, Lapalus M, Delaunay JL, Gonzales E, Jacquemin E, Aït-Slimane T, Falguières T. Int J Mol Sci. 2021 Feb 20;22(4):2113. doi: 10.3390/ijms22042113. PMID: 33672718.
- Effect of CFTR correctors on the traffic and the function of intracellularly retained ABCB4 variants. Ben Saad A, Vauthier V, Tóth Á, Janaszkiewicz A, Durand-Schneider AM, Bruneau A, Delaunay JL, Lapalus M, Mareux E, Garcin I, Gonzales E, Housset C, Aït-Slimane T, Jacquemin E, Di Meo F, Falguières T. Liver Int. 2021 Jun;41(6):1344-1357. doi: 10.1111/liv.14839. PMID: 33650203.
- [The EGFR ligand amphiregulin protects from cholestatic liver injury]. Sidibé N, Jalabert H, Merlen G, Tordjmann T. Rôle hépato-protecteur de l’amphiréguline, un ligand du récepteur de l’EGF, en situation de cholestase Med Sci (Paris). 2021 Jan;37(1):103-105. French. doi: 10.1051/medsci/2020265. PMID: 33492227.
- Pharmacological Premature Termination Codon Readthrough of ABCB11 in Bile Salt Export Pump Deficiency: An In Vitro Study. Amzal R, Thébaut A, Lapalus M, Almes M, Grosse B, Mareux E, Collado-Hilly M, Davit-Spraul A, Bidou L, Namy O, Jacquemin E, Gonzales E. Hepatology. 2021 Apr;73(4):1449-1463. doi: 10.1002/hep.31476. PMID: 32702170.
- Ursodeoxycholic acid therapy throughout pregnancy in women affected with chronic cholestasis of childhood: No evidence for teratogenicity. Lykavieris P, Bernard O, Jacquemin E. Clin Res Hepatol Gastroenterol. 2021 May;45(3):101472. doi: 10.1016/j.clinre.2020.05.020. PMID: 32565202.
- [Rituximab in the treatment of diffuse large B-cell lymphoma: The CACNA1C subunit of channel Cav1.2 expression linked to certain forms of resistance]. Bouabdallah S, Mariko ML, Besson J, Dellis O. Le rituximab dans le traitement du lymphome diffus à grandes cellules B – L’expression de la sous-unité CACNA1C du canal Cav 1.2 liée à certaines formes de résistance. Med Sci (Paris). 2021 Apr;37(4):406-408. French. doi: 10.1051/medsci/2021043. PMID: 33908862.
2020
- TGR5 controls bile acid composition and gallbladder function to protect the liver from bile acid overload. Bidault-Jourdainne V, Merlen G, Glénisson M, Doignon I, Garcin I, Péan N, Boisgard R, Ursic-Bedoya J, Serino M, Ullmer C, Humbert L, Abdelrafee A, Golse N, Vibert E, Duclos-Vallée JC, Rainteau D, Tordjmann T. JHEP Rep. 2020 Nov 11;3(2):100214. doi: 10.1016/j.jhepr.2020.100214. PMID: 33604531.
- Synthesis and Characterization of Store-Operated Calcium Entry Inhibitors Active in the Submicromolar Range. Le Guilcher C, Luyten T, Parys JB, Pucheault M, Dellis O. Int J Mol Sci. 2020 Dec 21;21(24):9777. doi: 10.3390/ijms21249777. PMID: 33371518.
- Generation and Quantitative Characterization of Functional and Polarized Biliary Epithelial Cysts. Bouzhir L, Gontran E, Loarca L, Collado-Hilly M, Dupuis-Williams P. J Vis Exp. 2020 May 16;(159). doi: 10.3791/61404. PMID: 32478726.
- Glycerol Phenylbutyrate Therapy in Progressive Familial Intrahepatic Cholestasis Type 2. Almes M, Jobert A, Lapalus M, Mareux E, Gonzales E, Jacquemin E. J Pediatr Gastroenterol Nutr. 2020 Jun;70(6):e139-e140. doi: 10.1097/MPG.0000000000002713. PMID: 32443059.
- Functional rescue of an ABCB11 mutant by ivacaftor: A new targeted pharmacotherapy approach in bile salt export pump deficiency. Mareux E, Lapalus M, Amzal R, Almes M, Aït-Slimane T, Delaunay JL, Adnot P, Collado-Hilly M, Davit-Spraul A, Falguières T, Callebaut I, Gonzales E, Jacquemin E. Liver Int. 2020 Aug;40(8):1917-1925. doi: 10.1111/liv.14518. PMID: 32433800.
- Hepatoprotective impact of the bile acid receptor TGR5. Merlen G, Bidault-Jourdainne V, Kahale N, Glenisson M, Ursic-Bedoya J, Doignon I, Garcin I, Humbert L, Rainteau D, Tordjmann T. Liver Int. 2020 May;40(5):1005-1015. doi: 10.1111/liv.14427. PMID: 32145703.
- Genotype correlates with the natural history of severe bile salt export pump deficiency. van Wessel DBE, Thompson RJ, Gonzales E, Jankowska I, Sokal E, Grammatikopoulos T, Kadaristiana A, Jacquemin E, Spraul A, Lipiński P, Czubkowski P, Rock N, Shagrani M, Broering D, Algoufi T, Mazhar N, Nicastro E, Kelly DA, Nebbia G, Arnell H, Björn Fischler, Hulscher JBF, Serranti D, Arikan C, Polat E, Debray D, Lacaille F, Goncalves C, Hierro L, Muñoz Bartolo G, Mozer-Glassberg Y, Azaz A, Brecelj J, Dezsőfi A, Calvo PL, Grabhorn E, Sturm E, van der Woerd WJ, Kamath BM, Wang JS, Li L, Durmaz Ö, Onal Z, Bunt TMG, Hansen BE, Verkade HJ; NAtural course and Prognosis of PFIC and Effect of biliary Diversion (NAPPED) consortium. J Hepatol. 2020 Jul;73(1):84-93. doi: 10.1016/j.jhep.2020.02.007. PMID: 32087350.
- A Novel CFC1 Mutation in a Family With Heterotaxy and Biliary Atresia Splenic Malformation Syndromes. Gonzales E, Davit-Spraul A, Jacquemin E. J Pediatr Gastroenterol Nutr. 2020 Jan;70(1):e24-e25. doi: 10.1097/MPG.0000000000002531. PMID: 31633655.
- TGR5-dependent hepatoprotection through the regulation of biliary epithelium barrier function. Merlen G, Kahale N, Ursic-Bedoya J, Bidault-Jourdainne V, Simerabet H, Doignon I, Tanfin Z, Garcin I, Péan N, Gautherot J, Davit-Spraul A, Guettier C, Humbert L, Rainteau D, Ebnet K, Ullmer C, Cassio D, Tordjmann T. Gut. 2020 Jan;69(1):146-157. doi: 10.1136/gutjnl-2018-316975. PMID: 30723104.
2019
- Biophysical Control of Bile Duct Epithelial Morphogenesis in Natural and Synthetic Scaffolds. Funfak A, Bouzhir L, Gontran E, Minier N, Dupuis-Williams P, Gobaa S. Front Bioeng Biotechnol. 2019 Dec 13;7:417. doi: 10.3389/fbioe.2019.00417. PMID: 31921820.
- Improvement of the rituximab-induced cell death by potentiation of the store-operated calcium entry in mantle cell lymphoma cell lines. Doignon I, Fayol O, Dellis O. Oncotarget. 2019 Jul 9;10(43):4466-4478. doi: 10.18632/oncotarget.27063. PMID: 31320998.
- Structural analogues of roscovitine rescue the intracellular traffic and the function of ER-retained ABCB4 variants in cell models. Vauthier V, Ben Saad A, Elie J, Oumata N, Durand-Schneider AM, Bruneau A, Delaunay JL, Housset C, Aït-Slimane T, Meijer L, Falguières T. Sci Rep. 2019 Apr 30;9(1):6653. doi: 10.1038/s41598-019-43111-y. PMID: 31040306.
2018
- Cholic acid for primary bile acid synthesis defects: a life-saving therapy allowing a favorable outcome in adulthood. Gonzales E, Matarazzo L, Franchi-Abella S, Dabadie A, Cohen J, Habes D, Hillaire S, Guettier C, Taburet AM, Myara A, Jacquemin E. Orphanet J Rare Dis. 2018 Oct 29;13(1):190. doi: 10.1186/s13023-018-0920-5. PMID: 30373615.
- The P2X4 purinergic receptor regulates hepatic myofibroblast activation during liver fibrogenesis. Le Guilcher C, Garcin I, Dellis O, Cauchois F, Tebbi A, Doignon I, Guettier C, Julien B, Tordjmann T. J Hepatol. 2018 Sep;69(3):644-653. doi: 10.1016/j.jhep.2018.05.020. PMID: 29802948.
- Morphological characterization of chronic antibody-mediated rejection in ABO-identical or ABO-compatible pediatric liver graft recipients. Dao M, Habès D, Taupin JL, Mussini C, Redon MJ, Suberbielle C, Jacquemin E, Gonzales E, Guettier C. Liver Transpl. 2018 Jul;24(7):897-907. doi: 10.1002/lt.25187. PMID: 29704327.
2017
- [Gal-9 promotes viral persistence of hepatitis virus in the liver]. Zahaf A, Badia A, Morel J, Dellis O. La galectine-9 favorise la persistance du virus de l’hépatite C dans le foie. Med Sci (Paris). 2017 Nov;33(11):947-949. French. doi: 10.1051/medsci/20173311010. PMID: 29200391.
- [Cholestasis-induced liver injury: the role of S1PR2]. Dissous L, Cesard E, Dance A, Tordjmann T. Les lésions hépatiques induites par la cholestase : le rôle de S1PR2. Med Sci (Paris). 2017 Jun-Jul;33(6-7):606-609. French. doi: 10.1051/medsci/20173306016. PMID: 28990561.
- Mutations in the novel gene FOPV are associated with familial autosomal dominant and non-familial obliterative portal venopathy. Besmond C, Valla D, Hubert L, Poirier K, Grosse B, Guettier C, Bernard O, Gonzales E, Jacquemin E. Liver Int. 2018 Feb;38(2):358-364. doi: 10.1111/liv.13547. PMID: 28792652.
- Cholic Acid to Treat HSD3B7 and AKR1D1 Deficiencies. Jacquemin E, Gonzales E. J Pediatr Gastroenterol Nutr. 2017 Dec;65(6):e134. doi: 10.1097/MPG.0000000000001693. PMID: 28727657.
- Bile acids and their receptors during liver regeneration: « Dangerous protectors ». Merlen G, Ursic-Bedoya J, Jourdainne V, Kahale N, Glenisson M, Doignon I, Rainteau D, Tordjmann T. Mol Aspects Med. 2017 Aug;56:25-33. doi: 10.1016/j.mam.2017.03.002. PMID: 28302491.
- Attempt to Determine the Prevalence of Two Inborn Errors of Primary Bile Acid Synthesis: Results of a European Survey. Jahnel J, Zöhrer E, Fischler B, D’Antiga L, Debray D, Dezsofi A, Haas D, Hadzic N, Jacquemin E, Lamireau T, Maggiore G, McKiernan PJ, Calvo PL, Verkade HJ, Hierro L, McLin V, Baumann U, Gonzales E. J Pediatr Gastroenterol Nutr. 2017 Jun;64(6):864-868. doi: 10.1097/MPG.0000000000001546. PMID: 28267072.
- Targeted pharmacotherapies for defective ABC transporters. Vauthier V, Housset C, Falguières T. Biochem Pharmacol. 2017 Jul 15;136:1-11. doi: 10.1016/j.bcp.2017.02.020. PMID: 28245962.
- Sertraline as an Additional Treatment for Cholestatic Pruritus in Children. Thébaut A, Habes D, Gottrand F, Rivet C, Cohen J, Debray D, Jacquemin E, Gonzales E. J Pediatr Gastroenterol Nutr. 2017 Mar;64(3):431-435. doi: 10.1097/MPG.0000000000001385. PMID: 27557426.
- MYO5B mutations cause cholestasis with normal serum gamma-glutamyl transferase activity in children without microvillous inclusion disease. Gonzales E, Taylor SA, Davit-Spraul A, Thébaut A, Thomassin N, Guettier C, Whitington PF, Jacquemin E. Hepatology. 2017 Jan;65(1):164-173. doi: 10.1002/hep.28779. PMID: 27532546.
2016
- Oral Tocofersolan Corrects or Prevents Vitamin E Deficiency in Children With Chronic Cholestasis. Thébaut A, Nemeth A, Le Mouhaër J, Scheenstra R, Baumann U, Koot B, Gottrand F, Houwen R, Monard L, de Micheaux SL, Habes D, Jacquemin E. J Pediatr Gastroenterol Nutr. 2016 Dec;63(6):610-615. doi: 10.1097/MPG.0000000000001331. PMID: 27429423.
- [Cholangiocyte proliferation induced by bile acids: impact of TGR5]. Barichon C, Correia C, Tordjmann T. La prolifération des cholangiocytes induite par les acides biliaires : place du récepteur TGR5. Med Sci (Paris). 2016 Jun-Jul;32(6-7):585-7. French. doi: 10.1051/medsci/20163206020. PMID: 27406767.
- The P2X4 purinergic receptor impacts liver regeneration after partial hepatectomy in mice through the regulation of biliary homeostasis. Besnard A, Gautherot J, Julien B, Tebbi A, Garcin I, Doignon I, Péan N, Gonzales E, Cassio D, Grosse B, Liu B, Safya H, Cauchois F, Humbert L, Rainteau D, Tordjmann T. Hepatology. 2016 Sep;64(3):941-53. doi: 10.1002/hep.28675. PMID: 27301647.
- Cholestasis Reveals Severe Cortisol Deficiency in Neonatal Pituitary Stalk Interruption Syndrome. Mauvais FX, Gonzales E, Davit-Spraul A, Jacquemin E, Brauner R. PLoS One. 2016 Feb 1;11(2):e0147750. doi: 10.1371/journal.pone.0147750. PMID: 26829045.
- Serum Autotaxin Activity Correlates With Pruritus in Pediatric Cholestatic Disorders. Kremer AE, Gonzales E, Schaap FG, Oude Elferink RP, Jacquemin E, Beuers U. J Pediatr Gastroenterol Nutr. 2016 Apr;62(4):530-5. doi: 10.1097/MPG.0000000000001044. PMID: 26628447.
- A functional classification of ABCB4 variations causing progressive familial intrahepatic cholestasis type 3. Delaunay JL, Durand-Schneider AM, Dossier C, Falguières T, Gautherot J, Davit-Spraul A, Aït-Slimane T, Housset C, Jacquemin E, Maurice M. Hepatology. 2016 May;63(5):1620-31. doi: 10.1002/hep.28300. PMID: 26474921.
2015
- The Bile Acid Receptor TGR5 and Liver Regeneration. Jourdainne V, Péan N, Doignon I, Humbert L, Rainteau D, Tordjmann T. Dig Dis. 2015;33(3):319-26. doi: 10.1159/000371668. PMID: 26045264.
- Potentiation of the store-operated calcium entry (SOCE) induces phytohemagglutinin-activated Jurkat T cell apoptosis. Djillani A, Doignon I, Luyten T, Lamkhioued B, Gangloff SC, Parys JB, Nüße O, Chomienne C, Dellis O. Cell Calcium. 2015 Aug;58(2):171-85. doi: 10.1016/j.ceca.2015.04.005. PMID: 25963393.
- Targeted pharmacotherapy in progressive familial intrahepatic cholestasis type 2: Evidence for improvement of cholestasis with 4-phenylbutyrate. Gonzales E, Grosse B, Schuller B, Davit-Spraul A, Conti F, Guettier C, Cassio D, Jacquemin E. Hepatology. 2015 Aug;62(2):558-66. doi: 10.1002/hep.27767. PMID: 25716872.
- Bile acids and FGF receptors: orchestrators of optimal liver regeneration. Gilgenkrantz H, Tordjmann T. Gut. 2015 Sep;64(9):1351-2. doi: 10.1136/gutjnl-2014-308746. PMID: 25654989.
- Paramecium swimming and ciliary beating patterns: a study on four RNA interference mutations. Funfak A, Fisch C, Abdel Motaal HT, Diener J, Combettes L, Baroud CN, Dupuis-Williams P. Integr Biol (Camb). 2015 Jan;7(1):90-100. doi: 10.1039/c4ib00181h. PMID: 25383612.
2014
- Liver transcript analysis reveals aberrant splicing due to silent and intronic variations in the ABCB11 gene. Davit-Spraul A, Oliveira C, Gonzales E, Gaignard P, Thérond P, Jacquemin E. Mol Genet Metab. 2014 Nov;113(3):225-9. doi: 10.1016/j.ymgme.2014.07.006. PMID: 25085279.
- ABCB4: Insights from pathobiology into therapy. Falguières T, Aït-Slimane T, Housset C, Maurice M. Clin Res Hepatol Gastroenterol. 2014 Oct;38(5):557-63. doi: 10.1016/j.clinre.2014.03.001. PMID: 24953525.
- Phosphorylation of ABCB4 impacts its function: insights from disease-causing mutations. Gautherot J, Delautier D, Maubert MA, Aït-Slimane T, Bolbach G, Delaunay JL, Durand-Schneider AM, Firrincieli D, Barbu V, Chignard N, Housset C, Maurice M, Falguières T. Hepatology. 2014 Aug;60(2):610-21. doi: 10.1002/hep.27170. PMID: 24723470.
- Characterization of novel store-operated calcium entry effectors. Djillani A, Nüße O, Dellis O. Biochim Biophys Acta. 2014 Oct;1843(10):2341-7. doi: 10.1016/j.bbamcr.2014.03.012. PMID: 24657813.
- Secondary Mitochondrial Respiratory Chain Defect Can Delay Accurate PFIC2 Diagnosis. Davit-Spraul A, Beinat M, Debray D, Rötig A, Slama A, Jacquemin E. JIMD Rep. 2014;14:17-21. doi: 10.1007/8904_2013_278. Epub 2013 Nov 9. PMID: 24214725.
- Clinical utility gene card for: progressive familial intrahepatic cholestasis type 3. Gonzales E, Spraul A, Jacquemin E. Eur J Hum Genet. 2014 Apr;22(4). doi: 10.1038/ejhg.2013.188. PMID: 24002166.
- Clinical utility gene card for: progressive familial intrahepatic cholestasis type 2. Gonzales E, Spraul A, Jacquemin E. Eur J Hum Genet. 2014 Apr;22(4). doi: 10.1038/ejhg.2013.187. PMID: 23982689.
- Clinical utility gene card for: progressive familial intrahepatic cholestasis type 1. Gonzales E, Spraul A, Jacquemin E. Eur J Hum Genet. 2014 Apr;22(4). doi: 10.1038/ejhg.2013.186. PMID: 23982690.
- Gut microbiota and bile acids: an old story revisited (again). Péan N, Doignon I, Tordjmann T. Clin Res Hepatol Gastroenterol. 2014 Apr;38(2):129-31. doi: 10.1016/j.clinre.2013.06.006. PMID: 23916556.
- The receptor TGR5 protects the liver from bile acid overload during liver regeneration in mice. Péan N, Doignon I, Garcin I, Besnard A, Julien B, Liu B, Branchereau S, Spraul A, Guettier C, Humbert L, Schoonjans K, Rainteau D, Tordjmann T. Hepatology. 2013 Oct;58(4):1451-60. doi: 10.1002/hep.26463. PMID: 23686672.
- Arthrogryposis, renal dysfunction, and cholestasis syndrome caused by VIPAR mutation. Ackermann O, Gonzales E, Keller M, Guettier C, Gissen P, Jacquemin E. J Pediatr Gastroenterol Nutr. 2014 Mar;58(3):e29-32. doi: 10.1097/MPG.0b013e318298108f. PMID: 23636179.
Collaborations and partnerships
Under construction...
Our funders




