
Equipe 4 – Thierry Tordjmann
Homéostasie biliaire et réparation du foie
Activités de Recherche
1. Homéostasie biliaire et réparation hépatique
Responsable : Dr Thierry Tordjmann
Même si la capacité de régénération du foie est importante, la réparation tissulaire lors de lésions chroniques (d’étiologies biliaires ou non biliaires) peut entraîner une fibrose hépatique. Il est important de noter qu’après une blessure et pendant la réparation du foie, les fonctions hépatiques doivent être maintenues pour assurer la demande périphérique. Ceci est particulièrement critique pour la sécrétion de la bile, qui doit être finement réglée afin de préserver le parenchyme hépatique d’une blessure induite par les acides biliaires. Cependant, les mécanismes permettant au foie de maintenir l’homéostasie biliaire pendant la réparation après une blessure ne sont pas complètement compris. Dans ce contexte, nos travaux sont axés sur les relations mutuelles entre l’homéostasie biliaire et la réparation du foie. Une attention particulière est portée aux acides biliaires et à leurs récepteurs (notamment TGR5) comme cibles thérapeutiques potentielles. A cette fin, nous utilisons des modèles in vivo (souris) de surcharge en acides biliaires et in vitro (culture d’hépatocytes et de cholangiocytes), ainsi que des échantillons hépatiques humains (tissus et cellules isolées).
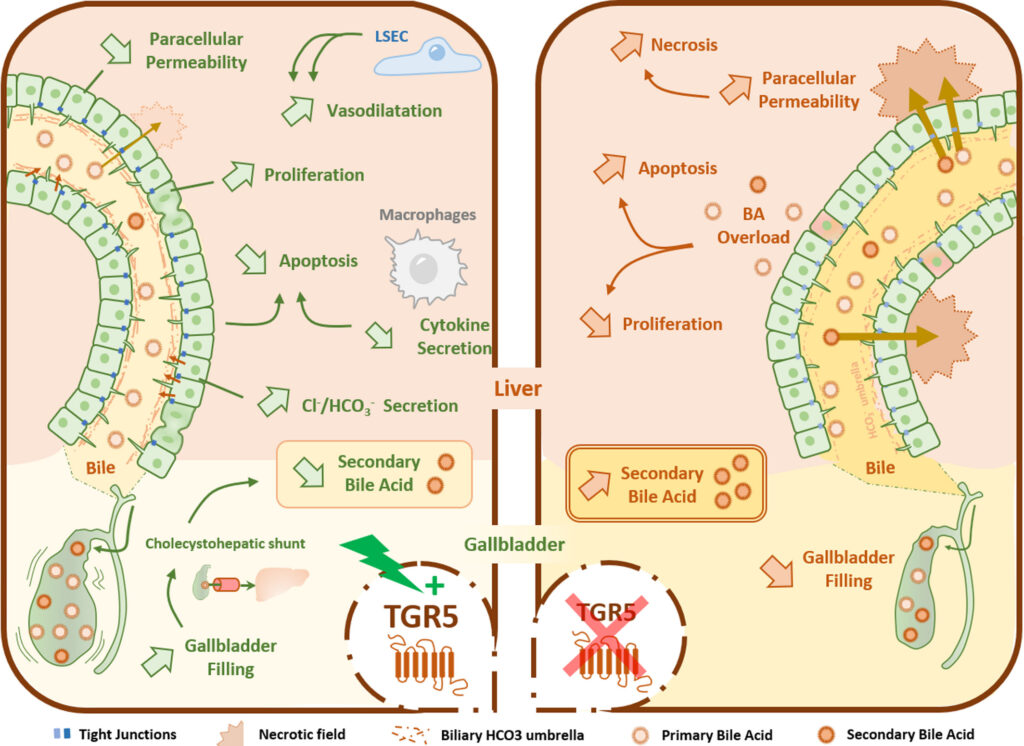
Figure : Représentation schématique des différents mécanismes d’hépato-protection médiés par TGR5. L’intégrité de la barrière hémato-biliaire (BHE), qui sépare le sang de la bile, est essentielle dans les conditions physiologiques et pathologiques. La stimulation du récepteur TGR5 favorise la prolifération des cholangiocytes et la fonction de barrière en renforçant les jonctions serrées des cholangiocytes. TGR5 régule le transport des ions chlorure et bicarbonate dans la bile, réduisant ainsi la protonation des acides biliaires et protégeant le parenchyme hépatique de la cytotoxicité des mêmes acides biliaires. TGR5 a un impact physiologique sur les fonctions de la vésicule biliaire, y compris sur la réabsorption des acides biliaires identifiée comme le shunt cholécysto-hépatique, limitant la quantité d’acides biliaires secondaires toxiques. La signalisation médiée par les acides biliaires via TGR5 réduit le tonus vasculaire hépatique et la pression portale, résultant d’une vasodilatation sinusoïdale (d’après Merlen et al., Liver Int 2020;40(5):1005-1015).
2. Physiopathologie et traitement des cholestases génétiques
Responsable : Pr Emmanuel Jacquemin
Les maladies hépatiques cholestatiques chroniques peuvent avoir des origines génétiques en lien avec des défauts moléculaires des transporteurs ABC (ATP-Binding-Cassette) localisés au niveau de la membrane canaliculaire des hépatocytes. En effet, les variations génétiques d’ABCB11/BSEP (transporteur des acides biliaires) et d’ABCB4/MDR3 (transporteur de phospholipides) sont impliquées dans des formes rares et sévères de cholestases génétiques appelées cholestases familiales intrahépatiques progressives de types 2 et 3, respectivement. Ces maladies se caractérisent par une cholestase (arrêt ou diminution de l’écoulement de la bile), un ictère (jaunisse) et un prurit (démangeaisons) modéré à sévère, et évoluent le plus souvent vers une insuffisance hépatique, nécessitant alors une transplantation hépatique. L’objectif de nos travaux est d’identifier de nouvelles molécules capables de corriger les défauts des transporteurs ABC canaliculaires mutés (expression, trafic ou fonction), dans le cadre de ces maladies cholestatiques. Pour cela, nous utilisons des modèles cellulaires exprimant les transporteurs ABC canaliculaires défectueux afin d’identifier des molécules d’intérêt thérapeutique capables de restaurer leur synthèse, leur trafic intracellulaire et/ou leur fonction. A terme, les molécules les plus pertinentes seront validées à un stade préclinique dans des modèles murins mimant les pathologies humaines avant d’envisager leur transfert auprès des patients dans une perspective de médecine personnalisée.
Figure : Représentation schématique des classes de variants génétiques des transporteurs ABC canaliculaires. Les classes ont été déterminées en fonction du déficit identifié dans des modèles cellulaires. La classe I correspond à des variants non-sens (codon stop prématuré) aboutissant à une expression protéique extrêmement réduite, voire nulle. Les variants de classe II, III et IV sont principalement des variants faux-sens (remplacement d’un acide aminé par un autre) dont le déficit est lié à des défauts de trafic intracellulaire, d’activité ou de stabilité, respectivement. Les variants de classe V ne présentent aucun défaut apparent, identifiable par les approches expérimentales utilisées et pourraient correspondre à des polymorphismes non délétères (d’après Vauthier et al., Biochem Pharmacol 2017;136:1-11).
3. Mécanismes moléculaires des ciliopathies et organogénèse biliaire
Responsable : Dr Pascale Dupuis-Williams
La construction au cours de l’embryogenèse et le maintien d’un système biliaire fonctionnel impliquent notamment le cil primaire, dont des dysfonctions sont associées à des ciliopathies biliaires. Bien que de nombreux travaux aient permis d’élucider le rôle du cil primaire des cholangiocytes dans la signalisation et la régulation de l’homéostasie biliaire, sa fonction dans l’organogénèse biliaire reste à élucider. Dans ce contexte, nous développons nos travaux selon deux axes : i) un programme de recherche destiné à décrypter le rôle des protéines du cil primaire dans la polarité épithéliale des cholangiocytes ; et ii) une activité de bioingénierie visant à la construction de tubes biliaires pour fournir des modèles de développement des voies biliaires in vitro et des outils pour la médecine régénérative.
Membres de l’équipe
CHERCHEURS ET ENSEIGNANTS-CHERCHEURS

Olivier DELLIS
Maître de Conférence
Université Paris-Saclay
01-69-15-49-59
olivier.dellis@universite-paris-saclay.fr

Pascale DUPUIS-WILLIAMS
Maître de Conférence
ESPCI Paris
01-69-15-68-66
pascale.dupuis-williams@universite-paris-saclay.fr

Thomas FALGUIERES
Chargé de Recherche
Inserm
01-69-15-62-94
thomas.falguieres@inserm.fr

Grégory MERLEN
Chargé de Recherche
Inserm
01-69-15-68-59
gregory.merlen@inserm.fr

Thierry TORDJMANN
Directeur de Recherche
Inserm
01-69-15-68-63
thierry.tordjmann@universite-paris-saclay.fr
CLINICIENS
Oanez ACKERMANN
PH
AP-HP
01-45-21-31-67
oanez.ackermann@aphp.fr

Marion ALMES
CCA
AP-HP / Université-Paris-Saclay
01-45-21-31-67
marionflorence.almes@aphp.fr

Emmanuel GONZALES
PU-PH
AP-HP / Université-Paris-Saclay
01-45-21-31-67
emmanuel.gonzales@aphp.fr
Dalila HABES
PH
AP-HP
01-45-21-31-67
dalila.habes@aphp.fr
Bogdan HERMEZIU
PH
AP-HP
01-45-21-31-67
bogdan.hermeziu@aphp.fr

Emmanuel JACQUEMIN
PU-PH
AP-HP / Université-Paris-Saclay
01-45-21-31-67
emmanuel.jacquemin@aphp.fr
Anne SPRAUL
PH
AP-HP
01-45-21-35-22
anne.spraul@aphp.fr
Alice THEBAUT
PH
AP-HP
01-45-21-31-67
alice.thebaut@aphp.fr
INGENIEURS ET TECHNICIENS

Manon BANET
Ingénieure d’Etude (contract.)
Inserm (ANR)
01-69-15-62-94
manon.banet@inserm.fr

Latifa BOUZHIR
Ingénieure d’Etude
Inserm
01-69-15-68-56
latifa.bouzhir@universite-paris-saclay.fr

Khadidiatou DIARRA
Adjoint technique
Inserm
01-69-15-68-58
khadidiatou.diarra@inserm.fr

Isabelle DOIGNON
Ingénieure d’Etude
Inserm
01-69-15-68-64
isabelle.doignon@universite-paris-saclay.fr

Olivier FAYOL
Technicien
Université Paris-Saclay
01-69-15-63-96
olivier.fayol@universite-paris-saclay.fr

Isabelle GARCIN
Ingénieure d’Etude
Université Paris-Saclay
01-69-15-76-39
isabelle.garcin@universite-paris-saclay.fr

Martine LAPALUS
Ingénieure de Recherche
Inserm
01-69-15-39-25
martine.lapalus@universite-paris-saclay.fr
DOCTORANTS ET POST-doctorants

Chloé CAENEN
Doctorante
Université Paris-Saclay
01-69-15-58-24
chloe.caenen@inserm.fr

Mounia LAKLI
Doctorante
Université Paris-Saclay
01-69-15-62-94
mounia.lakli@inserm.fr

Thibault PEBRIER
Doctorant
Université Paris-Saclay
01-69-15-63-96
thibault.pebrier@universite-paris-saclay.fr
ANCIENS MEMBRES DE L’éQUIPE
XXX XXXXX
XXXX
XXXXXXXXXX
01-XX-XX-XX-XX
xxxx.xxxxx@xxxx.com
XXX XXXXX
XXXX
XXXXXXXXXX
01-XX-XX-XX-XX
xxxx.xxxxx@xxxx.com
XXX XXXXX
XXXX
XXXXXXXXXX
01-XX-XX-XX-XX
xxxx.xxxxx@xxxx.com
Publications principales
2022
- In Vitro Rescue of the Bile Acid Transport Function of ABCB11 Variants by CFTR Potentiators. Mareux E, Lapalus M, Ben Saad A, Zelli R, Lakli M, Riahi Y, Almes M, Banet M, Callebaut I, Decout JL, Falguières T, Jacquemin E, Gonzales E. Int J Mol Sci. 2022 Sept 15;23(18):10758. doi:10.3390/ijms231810758.
- ABC Transporters in Human Diseases: Future Directions and Therapeutic Perspectives. Falguières T. Int J Mol Sci. 2022 Apr 12;23(8):4250. doi: 10.3390/ijms23084250. PMID: 35457067.
2021
- Construction of functional biliary epithelial branched networks with predefined geometry using digital light stereolithography. Mazari-Arrighi E, Ayollo D, Farhat W, Marret A, Gontran E, Dupuis-Williams P, Larghero J, Chatelain F, Fuchs A. Biomaterials. 2021 Dec;279:121207. doi: 10.1016/j.biomaterials.2021.121207. PMID: 34741977.
- Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): a randomised phase 2 study. Gonzales E, Hardikar W, Stormon M, Baker A, Hierro L, Gliwicz D, Lacaille F, Lachaux A, Sturm E, Setchell KDR, Kennedy C, Dorenbaum A, Steinmetz J, Desai NK, Wardle AJ, Garner W, Vig P, Jaecklin T, Sokal EM, Jacquemin E. Lancet. 2021 Oct 30;398(10311):1581-1592. doi: 10.1016/S0140-6736(21)01256-3. PMID: 34755627.
- In vitro functional rescue by ivacaftor of an ABCB11 variant involved in PFIC2 and intrahepatic cholestasis of pregnancy. Mareux E, Lapalus M, Ben-Saad A, Callebaut I, Falguières T, Gonzales E, Jacquemin E. Orphanet J Rare Dis. 2021 Nov 18;16(1):484. doi: 10.1186/s13023-021-02125-4. PMID: 34794484.
- Self-Organogenesis from 2D Micropatterns to 3D Biomimetic Biliary Trees. Gontran E, Loarca L, El Kassis C, Bouzhir L, Ayollo D, Mazari-Arrighi E, Fuchs A, Dupuis-Williams P. Bioengineering (Basel). 2021 Aug 5;8(8):112. doi: 10.3390/bioengineering8080112. PMID: 34436115.
- RAB10 Interacts with ABCB4 and Regulates Its Intracellular Traffic. Ben Saad A, Vauthier V, Lapalus M, Mareux E, Bennana E, Durand-Schneider AM, Bruneau A, Delaunay JL, Gonzales E, Housset C, Aït-Slimane T, Guillonneau F, Jacquemin E, Falguières T. Int J Mol Sci. 2021 Jun 30;22(13):7087. doi: 10.3390/ijms22137087. PMID: 34209301.
- Molecular Regulation of Canalicular ABC Transporters. Ben Saad A, Bruneau A, Mareux E, Lapalus M, Delaunay JL, Gonzales E, Jacquemin E, Aït-Slimane T, Falguières T. Int J Mol Sci. 2021 Feb 20;22(4):2113. doi: 10.3390/ijms22042113. PMID: 33672718.
- Effect of CFTR correctors on the traffic and the function of intracellularly retained ABCB4 variants. Ben Saad A, Vauthier V, Tóth Á, Janaszkiewicz A, Durand-Schneider AM, Bruneau A, Delaunay JL, Lapalus M, Mareux E, Garcin I, Gonzales E, Housset C, Aït-Slimane T, Jacquemin E, Di Meo F, Falguières T. Liver Int. 2021 Jun;41(6):1344-1357. doi: 10.1111/liv.14839. PMID: 33650203.
- [The EGFR ligand amphiregulin protects from cholestatic liver injury]. Sidibé N, Jalabert H, Merlen G, Tordjmann T. Rôle hépato-protecteur de l’amphiréguline, un ligand du récepteur de l’EGF, en situation de cholestase Med Sci (Paris). 2021 Jan;37(1):103-105. French. doi: 10.1051/medsci/2020265. PMID: 33492227.
- Pharmacological Premature Termination Codon Readthrough of ABCB11 in Bile Salt Export Pump Deficiency: An In Vitro Study. Amzal R, Thébaut A, Lapalus M, Almes M, Grosse B, Mareux E, Collado-Hilly M, Davit-Spraul A, Bidou L, Namy O, Jacquemin E, Gonzales E. Hepatology. 2021 Apr;73(4):1449-1463. doi: 10.1002/hep.31476. PMID: 32702170.
- Ursodeoxycholic acid therapy throughout pregnancy in women affected with chronic cholestasis of childhood: No evidence for teratogenicity. Lykavieris P, Bernard O, Jacquemin E. Clin Res Hepatol Gastroenterol. 2021 May;45(3):101472. doi: 10.1016/j.clinre.2020.05.020. PMID: 32565202.
- [Rituximab in the treatment of diffuse large B-cell lymphoma: The CACNA1C subunit of channel Cav1.2 expression linked to certain forms of resistance]. Bouabdallah S, Mariko ML, Besson J, Dellis O. Le rituximab dans le traitement du lymphome diffus à grandes cellules B – L’expression de la sous-unité CACNA1C du canal Cav 1.2 liée à certaines formes de résistance. Med Sci (Paris). 2021 Apr;37(4):406-408. French. doi: 10.1051/medsci/2021043. PMID: 33908862.
2020
- TGR5 controls bile acid composition and gallbladder function to protect the liver from bile acid overload. Bidault-Jourdainne V, Merlen G, Glénisson M, Doignon I, Garcin I, Péan N, Boisgard R, Ursic-Bedoya J, Serino M, Ullmer C, Humbert L, Abdelrafee A, Golse N, Vibert E, Duclos-Vallée JC, Rainteau D, Tordjmann T. JHEP Rep. 2020 Nov 11;3(2):100214. doi: 10.1016/j.jhepr.2020.100214. PMID: 33604531.
- Synthesis and Characterization of Store-Operated Calcium Entry Inhibitors Active in the Submicromolar Range. Le Guilcher C, Luyten T, Parys JB, Pucheault M, Dellis O. Int J Mol Sci. 2020 Dec 21;21(24):9777. doi: 10.3390/ijms21249777. PMID: 33371518.
- Generation and Quantitative Characterization of Functional and Polarized Biliary Epithelial Cysts. Bouzhir L, Gontran E, Loarca L, Collado-Hilly M, Dupuis-Williams P. J Vis Exp. 2020 May 16;(159). doi: 10.3791/61404. PMID: 32478726.
- Glycerol Phenylbutyrate Therapy in Progressive Familial Intrahepatic Cholestasis Type 2. Almes M, Jobert A, Lapalus M, Mareux E, Gonzales E, Jacquemin E. J Pediatr Gastroenterol Nutr. 2020 Jun;70(6):e139-e140. doi: 10.1097/MPG.0000000000002713. PMID: 32443059.
- Functional rescue of an ABCB11 mutant by ivacaftor: A new targeted pharmacotherapy approach in bile salt export pump deficiency. Mareux E, Lapalus M, Amzal R, Almes M, Aït-Slimane T, Delaunay JL, Adnot P, Collado-Hilly M, Davit-Spraul A, Falguières T, Callebaut I, Gonzales E, Jacquemin E. Liver Int. 2020 Aug;40(8):1917-1925. doi: 10.1111/liv.14518. PMID: 32433800.
- Hepatoprotective impact of the bile acid receptor TGR5. Merlen G, Bidault-Jourdainne V, Kahale N, Glenisson M, Ursic-Bedoya J, Doignon I, Garcin I, Humbert L, Rainteau D, Tordjmann T. Liver Int. 2020 May;40(5):1005-1015. doi: 10.1111/liv.14427. PMID: 32145703.
- Genotype correlates with the natural history of severe bile salt export pump deficiency. van Wessel DBE, Thompson RJ, Gonzales E, Jankowska I, Sokal E, Grammatikopoulos T, Kadaristiana A, Jacquemin E, Spraul A, Lipiński P, Czubkowski P, Rock N, Shagrani M, Broering D, Algoufi T, Mazhar N, Nicastro E, Kelly DA, Nebbia G, Arnell H, Björn Fischler, Hulscher JBF, Serranti D, Arikan C, Polat E, Debray D, Lacaille F, Goncalves C, Hierro L, Muñoz Bartolo G, Mozer-Glassberg Y, Azaz A, Brecelj J, Dezsőfi A, Calvo PL, Grabhorn E, Sturm E, van der Woerd WJ, Kamath BM, Wang JS, Li L, Durmaz Ö, Onal Z, Bunt TMG, Hansen BE, Verkade HJ; NAtural course and Prognosis of PFIC and Effect of biliary Diversion (NAPPED) consortium. J Hepatol. 2020 Jul;73(1):84-93. doi: 10.1016/j.jhep.2020.02.007. PMID: 32087350.
- A Novel CFC1 Mutation in a Family With Heterotaxy and Biliary Atresia Splenic Malformation Syndromes. Gonzales E, Davit-Spraul A, Jacquemin E. J Pediatr Gastroenterol Nutr. 2020 Jan;70(1):e24-e25. doi: 10.1097/MPG.0000000000002531. PMID: 31633655.
- TGR5-dependent hepatoprotection through the regulation of biliary epithelium barrier function. Merlen G, Kahale N, Ursic-Bedoya J, Bidault-Jourdainne V, Simerabet H, Doignon I, Tanfin Z, Garcin I, Péan N, Gautherot J, Davit-Spraul A, Guettier C, Humbert L, Rainteau D, Ebnet K, Ullmer C, Cassio D, Tordjmann T. Gut. 2020 Jan;69(1):146-157. doi: 10.1136/gutjnl-2018-316975. PMID: 30723104.
2019
- Biophysical Control of Bile Duct Epithelial Morphogenesis in Natural and Synthetic Scaffolds. Funfak A, Bouzhir L, Gontran E, Minier N, Dupuis-Williams P, Gobaa S. Front Bioeng Biotechnol. 2019 Dec 13;7:417. doi: 10.3389/fbioe.2019.00417. PMID: 31921820.
- Improvement of the rituximab-induced cell death by potentiation of the store-operated calcium entry in mantle cell lymphoma cell lines. Doignon I, Fayol O, Dellis O. Oncotarget. 2019 Jul 9;10(43):4466-4478. doi: 10.18632/oncotarget.27063. PMID: 31320998.
- Structural analogues of roscovitine rescue the intracellular traffic and the function of ER-retained ABCB4 variants in cell models. Vauthier V, Ben Saad A, Elie J, Oumata N, Durand-Schneider AM, Bruneau A, Delaunay JL, Housset C, Aït-Slimane T, Meijer L, Falguières T. Sci Rep. 2019 Apr 30;9(1):6653. doi: 10.1038/s41598-019-43111-y. PMID: 31040306.
2018
- Cholic acid for primary bile acid synthesis defects: a life-saving therapy allowing a favorable outcome in adulthood. Gonzales E, Matarazzo L, Franchi-Abella S, Dabadie A, Cohen J, Habes D, Hillaire S, Guettier C, Taburet AM, Myara A, Jacquemin E. Orphanet J Rare Dis. 2018 Oct 29;13(1):190. doi: 10.1186/s13023-018-0920-5. PMID: 30373615.
- The P2X4 purinergic receptor regulates hepatic myofibroblast activation during liver fibrogenesis. Le Guilcher C, Garcin I, Dellis O, Cauchois F, Tebbi A, Doignon I, Guettier C, Julien B, Tordjmann T. J Hepatol. 2018 Sep;69(3):644-653. doi: 10.1016/j.jhep.2018.05.020. PMID: 29802948.
- Morphological characterization of chronic antibody-mediated rejection in ABO-identical or ABO-compatible pediatric liver graft recipients. Dao M, Habès D, Taupin JL, Mussini C, Redon MJ, Suberbielle C, Jacquemin E, Gonzales E, Guettier C. Liver Transpl. 2018 Jul;24(7):897-907. doi: 10.1002/lt.25187. PMID: 29704327.
2017
- [Gal-9 promotes viral persistence of hepatitis virus in the liver]. Zahaf A, Badia A, Morel J, Dellis O. La galectine-9 favorise la persistance du virus de l’hépatite C dans le foie. Med Sci (Paris). 2017 Nov;33(11):947-949. French. doi: 10.1051/medsci/20173311010. PMID: 29200391.
- [Cholestasis-induced liver injury: the role of S1PR2]. Dissous L, Cesard E, Dance A, Tordjmann T. Les lésions hépatiques induites par la cholestase : le rôle de S1PR2. Med Sci (Paris). 2017 Jun-Jul;33(6-7):606-609. French. doi: 10.1051/medsci/20173306016. PMID: 28990561.
- Mutations in the novel gene FOPV are associated with familial autosomal dominant and non-familial obliterative portal venopathy. Besmond C, Valla D, Hubert L, Poirier K, Grosse B, Guettier C, Bernard O, Gonzales E, Jacquemin E. Liver Int. 2018 Feb;38(2):358-364. doi: 10.1111/liv.13547. PMID: 28792652.
- Cholic Acid to Treat HSD3B7 and AKR1D1 Deficiencies. Jacquemin E, Gonzales E. J Pediatr Gastroenterol Nutr. 2017 Dec;65(6):e134. doi: 10.1097/MPG.0000000000001693. PMID: 28727657.
- Bile acids and their receptors during liver regeneration: « Dangerous protectors ». Merlen G, Ursic-Bedoya J, Jourdainne V, Kahale N, Glenisson M, Doignon I, Rainteau D, Tordjmann T. Mol Aspects Med. 2017 Aug;56:25-33. doi: 10.1016/j.mam.2017.03.002. PMID: 28302491.
- Attempt to Determine the Prevalence of Two Inborn Errors of Primary Bile Acid Synthesis: Results of a European Survey. Jahnel J, Zöhrer E, Fischler B, D’Antiga L, Debray D, Dezsofi A, Haas D, Hadzic N, Jacquemin E, Lamireau T, Maggiore G, McKiernan PJ, Calvo PL, Verkade HJ, Hierro L, McLin V, Baumann U, Gonzales E. J Pediatr Gastroenterol Nutr. 2017 Jun;64(6):864-868. doi: 10.1097/MPG.0000000000001546. PMID: 28267072.
- Targeted pharmacotherapies for defective ABC transporters. Vauthier V, Housset C, Falguières T. Biochem Pharmacol. 2017 Jul 15;136:1-11. doi: 10.1016/j.bcp.2017.02.020. PMID: 28245962.
- Sertraline as an Additional Treatment for Cholestatic Pruritus in Children. Thébaut A, Habes D, Gottrand F, Rivet C, Cohen J, Debray D, Jacquemin E, Gonzales E. J Pediatr Gastroenterol Nutr. 2017 Mar;64(3):431-435. doi: 10.1097/MPG.0000000000001385. PMID: 27557426.
- MYO5B mutations cause cholestasis with normal serum gamma-glutamyl transferase activity in children without microvillous inclusion disease. Gonzales E, Taylor SA, Davit-Spraul A, Thébaut A, Thomassin N, Guettier C, Whitington PF, Jacquemin E. Hepatology. 2017 Jan;65(1):164-173. doi: 10.1002/hep.28779. PMID: 27532546.
2016
- Oral Tocofersolan Corrects or Prevents Vitamin E Deficiency in Children With Chronic Cholestasis. Thébaut A, Nemeth A, Le Mouhaër J, Scheenstra R, Baumann U, Koot B, Gottrand F, Houwen R, Monard L, de Micheaux SL, Habes D, Jacquemin E. J Pediatr Gastroenterol Nutr. 2016 Dec;63(6):610-615. doi: 10.1097/MPG.0000000000001331. PMID: 27429423.
- [Cholangiocyte proliferation induced by bile acids: impact of TGR5]. Barichon C, Correia C, Tordjmann T. La prolifération des cholangiocytes induite par les acides biliaires : place du récepteur TGR5. Med Sci (Paris). 2016 Jun-Jul;32(6-7):585-7. French. doi: 10.1051/medsci/20163206020. PMID: 27406767.
- The P2X4 purinergic receptor impacts liver regeneration after partial hepatectomy in mice through the regulation of biliary homeostasis. Besnard A, Gautherot J, Julien B, Tebbi A, Garcin I, Doignon I, Péan N, Gonzales E, Cassio D, Grosse B, Liu B, Safya H, Cauchois F, Humbert L, Rainteau D, Tordjmann T. Hepatology. 2016 Sep;64(3):941-53. doi: 10.1002/hep.28675. PMID: 27301647.
- Cholestasis Reveals Severe Cortisol Deficiency in Neonatal Pituitary Stalk Interruption Syndrome. Mauvais FX, Gonzales E, Davit-Spraul A, Jacquemin E, Brauner R. PLoS One. 2016 Feb 1;11(2):e0147750. doi: 10.1371/journal.pone.0147750. PMID: 26829045.
- Serum Autotaxin Activity Correlates With Pruritus in Pediatric Cholestatic Disorders. Kremer AE, Gonzales E, Schaap FG, Oude Elferink RP, Jacquemin E, Beuers U. J Pediatr Gastroenterol Nutr. 2016 Apr;62(4):530-5. doi: 10.1097/MPG.0000000000001044. PMID: 26628447.
- A functional classification of ABCB4 variations causing progressive familial intrahepatic cholestasis type 3. Delaunay JL, Durand-Schneider AM, Dossier C, Falguières T, Gautherot J, Davit-Spraul A, Aït-Slimane T, Housset C, Jacquemin E, Maurice M. Hepatology. 2016 May;63(5):1620-31. doi: 10.1002/hep.28300. PMID: 26474921.
2015
- The Bile Acid Receptor TGR5 and Liver Regeneration. Jourdainne V, Péan N, Doignon I, Humbert L, Rainteau D, Tordjmann T. Dig Dis. 2015;33(3):319-26. doi: 10.1159/000371668. PMID: 26045264.
- Potentiation of the store-operated calcium entry (SOCE) induces phytohemagglutinin-activated Jurkat T cell apoptosis. Djillani A, Doignon I, Luyten T, Lamkhioued B, Gangloff SC, Parys JB, Nüße O, Chomienne C, Dellis O. Cell Calcium. 2015 Aug;58(2):171-85. doi: 10.1016/j.ceca.2015.04.005. PMID: 25963393.
- Targeted pharmacotherapy in progressive familial intrahepatic cholestasis type 2: Evidence for improvement of cholestasis with 4-phenylbutyrate. Gonzales E, Grosse B, Schuller B, Davit-Spraul A, Conti F, Guettier C, Cassio D, Jacquemin E. Hepatology. 2015 Aug;62(2):558-66. doi: 10.1002/hep.27767. PMID: 25716872.
- Bile acids and FGF receptors: orchestrators of optimal liver regeneration. Gilgenkrantz H, Tordjmann T. Gut. 2015 Sep;64(9):1351-2. doi: 10.1136/gutjnl-2014-308746. PMID: 25654989.
- Paramecium swimming and ciliary beating patterns: a study on four RNA interference mutations. Funfak A, Fisch C, Abdel Motaal HT, Diener J, Combettes L, Baroud CN, Dupuis-Williams P. Integr Biol (Camb). 2015 Jan;7(1):90-100. doi: 10.1039/c4ib00181h. PMID: 25383612.
2014
- Liver transcript analysis reveals aberrant splicing due to silent and intronic variations in the ABCB11 gene. Davit-Spraul A, Oliveira C, Gonzales E, Gaignard P, Thérond P, Jacquemin E. Mol Genet Metab. 2014 Nov;113(3):225-9. doi: 10.1016/j.ymgme.2014.07.006. PMID: 25085279.
- ABCB4: Insights from pathobiology into therapy. Falguières T, Aït-Slimane T, Housset C, Maurice M. Clin Res Hepatol Gastroenterol. 2014 Oct;38(5):557-63. doi: 10.1016/j.clinre.2014.03.001. PMID: 24953525.
- Phosphorylation of ABCB4 impacts its function: insights from disease-causing mutations. Gautherot J, Delautier D, Maubert MA, Aït-Slimane T, Bolbach G, Delaunay JL, Durand-Schneider AM, Firrincieli D, Barbu V, Chignard N, Housset C, Maurice M, Falguières T. Hepatology. 2014 Aug;60(2):610-21. doi: 10.1002/hep.27170. PMID: 24723470.
- Characterization of novel store-operated calcium entry effectors. Djillani A, Nüße O, Dellis O. Biochim Biophys Acta. 2014 Oct;1843(10):2341-7. doi: 10.1016/j.bbamcr.2014.03.012. PMID: 24657813.
- Secondary Mitochondrial Respiratory Chain Defect Can Delay Accurate PFIC2 Diagnosis. Davit-Spraul A, Beinat M, Debray D, Rötig A, Slama A, Jacquemin E. JIMD Rep. 2014;14:17-21. doi: 10.1007/8904_2013_278. Epub 2013 Nov 9. PMID: 24214725.
- Clinical utility gene card for: progressive familial intrahepatic cholestasis type 3. Gonzales E, Spraul A, Jacquemin E. Eur J Hum Genet. 2014 Apr;22(4). doi: 10.1038/ejhg.2013.188. PMID: 24002166.
- Clinical utility gene card for: progressive familial intrahepatic cholestasis type 2. Gonzales E, Spraul A, Jacquemin E. Eur J Hum Genet. 2014 Apr;22(4). doi: 10.1038/ejhg.2013.187. PMID: 23982689.
- Clinical utility gene card for: progressive familial intrahepatic cholestasis type 1. Gonzales E, Spraul A, Jacquemin E. Eur J Hum Genet. 2014 Apr;22(4). doi: 10.1038/ejhg.2013.186. PMID: 23982690.
- Gut microbiota and bile acids: an old story revisited (again). Péan N, Doignon I, Tordjmann T. Clin Res Hepatol Gastroenterol. 2014 Apr;38(2):129-31. doi: 10.1016/j.clinre.2013.06.006. PMID: 23916556.
- The receptor TGR5 protects the liver from bile acid overload during liver regeneration in mice. Péan N, Doignon I, Garcin I, Besnard A, Julien B, Liu B, Branchereau S, Spraul A, Guettier C, Humbert L, Schoonjans K, Rainteau D, Tordjmann T. Hepatology. 2013 Oct;58(4):1451-60. doi: 10.1002/hep.26463. PMID: 23686672.
- Arthrogryposis, renal dysfunction, and cholestasis syndrome caused by VIPAR mutation. Ackermann O, Gonzales E, Keller M, Guettier C, Gissen P, Jacquemin E. J Pediatr Gastroenterol Nutr. 2014 Mar;58(3):e29-32. doi: 10.1097/MPG.0b013e318298108f. PMID: 23636179.
Collaborations et partenariats
En construction…
Nos financeurs




